1. 引言
维生素D的代谢研究证明:维生素D只有在人体内被转化为活性代谢物后才具有生理活性,代谢物的结构多为维生素D类的羟基衍生物,其中具有代表性的有:25-羟基维生素D3 (25-OH-VD3)、活性更强的1α,25-二羟基维生素D3 (1α,25-(OH)2-VD3) 1等。1α-羟基维生素D3 (1α-OH-VD3) 2虽不是维生素D3在人体内的直接代谢产物,但服用后即迅速转化为1,特殊的维生素D受体已经在三十多种人体组织中发现。因此维生素D类化合物也被应用当作灵敏的生物分子探针,用于分子生物学研究。活性维生素D3主要是1α,25-二羟基维生素D3,除了经典的体内钙磷调节功能外,近年来还发现它具有促进细胞分化和抑制细胞增殖的功能,可用于治疗恶性肿瘤如白血病、乳腺癌、子宫颈癌,皮肤增生性疾患如牛皮癣、脂溢性皮炎和免疫系统疾病如AIDS病等[1] [2] 。表1列出了制药公司开发成药物或正在临床试验的活性维生素D3类化合物的结构和名称。
人们在合成维生素D活性代谢产物如1α,25-二羟基维生素D3的同时,大量的类似物被合成. 在这些合成的类似物中,有的表现出更强的生理活性。如氟代的1α,25-二羟基维生素D3,其活性强度为原有母体分子的5~10倍,分子中含有环氧基团和过氧键侧链的化合物具有抑制骨髓白细胞增生的作用。1α,25-二羟基维生素D3在治疗剂量时,其钙活性可引起高钙症,使其应用受到限制。1990年Deluca等人[3] 发现,当1中A环19-亚甲基用两个氢取代时,形成的19-失碳-1α,25-二羟基维生素D3 3(图1),其细胞分化活性明显增加,而钙活性仅是1的十分之一,这一良好结果引起人们对1及其类似化合物合成的极大的兴趣。本文将对1α,25-二羟基维生素D3及其类似物的合成研究进展做一综述。
2. 1α,25-二羟基维生素D3的生物合成
天然维生素D类化合物是通过动物体或人体内的一些甾醇分子经脱氢和B环开裂转化而来的。如人体皮肤中含有的7-脱氢胆甾醇通过阳光中短紫外线的作用,发生周环反应,7-脱氢胆甾醇分子中的环己二烯开环转变成开链共轭三烯结构(即原维生素D3),第二步,在热的作用下,原维生素D3中相应的角甲基上一个ơ-H原子发生[1,7]迁移反应得维生素D3,无活性的维生素D3前体分子经肝脏转化为体内的主要循环形式25-羟基维生素D3,再经过肾脏转化为1α,25-二羟基维生素D3 1,通过这一活性形式调节各种生理功能[4] (Scheme 1)。
3. 1α,25-二羟基维生素D3及其类似物的合成研究进展
维生素D的一般合成策略是:首先合成由其分子中C5~C6键,或C6~C7键,或C7~C8键断裂形成的A环和带侧链的CD环,然后通过会聚合成连接A环和CD环。

Table 1. Some active vitamin D3 analogues as new drugs and new drug candidates
表1. 一些已经被开发成药物或正在临床试验的活性维生素D3类化合物

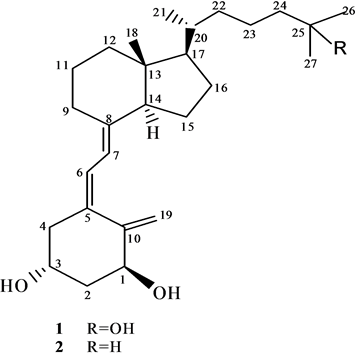
Figure 1. The chemical structures of 1α,25-dihydroxyvitamin D3 1 and 19-nor 1α, 25- dihydroxyvitamin D3 3
图1. 1α,25-二羟基维生素D3 1的和19-失碳-1α,25-二羟基维生素D3 3的化学结构

Scheme 1. Biosynthesis of 1α,25-dihydroxyvitamin D3
Scheme 1. 1α,25-二羟基维生素D3的生物合成途径
3.1. 通过C7~C8键连接A环和CD环的合成策略
3.1.1. Wittig-Horner法
1975年,Lythgoe等[5] 通过此方法,完成了维生素D的模型试验,反应中A 环连接的双键未发生迁移,新产生的C7~C8双键构型符合要求。此法成功地应用于1α,25-二羟基VD3的合成(Scheme 2)。
A环中间体4是维生素D全合成中使用频率最高的中间体. A环中间体4及其类似物可以通过不对称反应合成,也可以利用手性合成原料进行合成。Posner[6] 通过不对称的Diels-Alder反应以高收率和高e. e. 值合成得到环加成产物8, 8再经过开环﹑除砜基团﹑双键异构化﹑脱保护基和羟基保护以82%的总收率给出环己烯基羧酸酯10, 10经DIBAL还原、[3,3]σ迁移和异构化反应以约80%的收率给出12,然后被转化为4 (Scheme 3)。
Kabat[7] 从吡喃酮通过不对称的Ene反应经过16步反应以7%的总收率完成了18的合成(Scheme 4)。18是合成化合物4的前体。
Roumen等[8] 以(S)-香芹酮为原料,以合成路线合成了4(Scheme 5)。
另一个构建A环合成子的方法是由Takahashi[9] 开发的钯催化下的Heck反应,可用于合成1α,25-二羟基VD3的2-羟基衍生物A环合成子。它可由D-mannitol 21的三丙酮缩酮衍生物为原料合成。其合成路线如Scheme 6。
Kobayashi等[10] 以手性单酯27为原料,通过酶法在区域选择性和立体选择性的控制下合成了A环合成子4的前体31(Scheme 7)。
Ogasawara及其合作者[11] 发现了一个合成两种对映异构体形式的光学纯的三环二烯酮34,通过应用脂肪酶催化不对称合成内消旋前体,然后经过钯类催化剂对手性的单乙酰产物进行消除而得到。通过这个方法,他们又发现了一个新的合成A环合成子的方法。这样以(-)-34为原料经过10步反应,可以得到A环合成前体37,总收率为25%(Scheme 8)。
在合成1α,25-二羟基VD3类似物时,Deluca等[12] [13] 以(-)-奎尼酸为原料利用Wittig-Horner法合成了一系列新的2α或2β-羟基,2-甲基、2-亚甲基的1α,25-二羟基-19-失碳VD3的类似物。Yagamare [14] Sarandeses等[15] 采用Wittig-Horner法合成了CD环上带环状侧链结构的新的1α,25-二羟基VD3类似物。2010年Gleason等[16] 采用类似方法设计合成了双重官能团的邻氨基酰苯胺1α,25-二羟基维生素D3
类似物。它是一个强VDR拮抗剂。2013年Maehr等[17] 合成了重氢同位素标记的1α,25-二羟基维生素D3类似物,以研究其抗肿瘤活性及其体内代谢机理。
3.1.2. Cross-Coupling法
此法是利用金属试剂的偶联反应以C7~C8键连接方式构成维生素D类化合物,最早由Mourino和
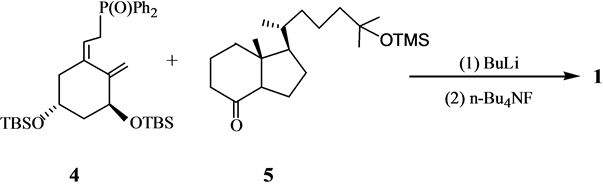
Scheme 2. Synthesis of 1α,25-dihydroxyvitamin D3 by Wittig-Horner reaction
Scheme 2. 1α,25-二羟基维生素D3的Wittig-Horner法合成
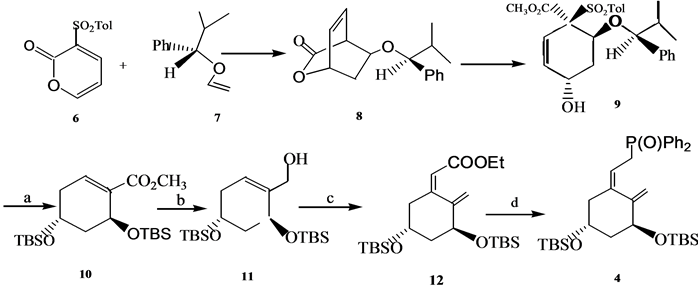
(a) 82%产率;(b) DIBAL;(c) PhS(O)CH2C(OEt)3,hv,9-fluorenone,89%; (d) (I) i-Bu2ALH,(II) NCS/Me2S,(III) Ph2PLi,(IV) 5% H2O2
Scheme 3. Synthesis of 1α,25-dihydroxyvitamin D3 A ring synthon 4
Scheme 3. 1α,25-二羟基维生素D3A环合成子4的合成

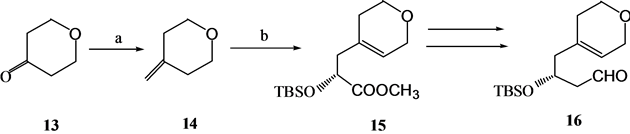
(a) Ph3 P =CH2; (b) OHCCOOCH3, (R)-Binol; (c) CH3Al(OCH3)Cl, CrO3
Scheme 4. Synthesis of 1α,25-dihydroxyvitamin D3 A ring synthon 18
Scheme 4. 1α,25-二羟基维生素D3 A环合成子18的合成
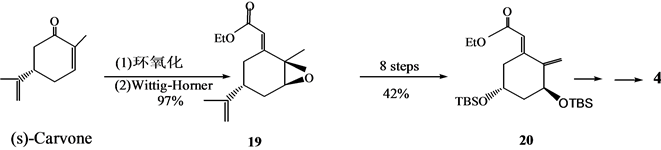
Scheme 5. Another synthesis of 1α,25-dihydroxyvitamin D3 A ring synthon 4
Scheme 5. 1α,25-二羟基维生素D3 A环合成子4的另法合成

Scheme 6. Synthesis of 1α,25-dihydroxyvitamin D3 A ring synthon 26
Scheme 6. 1α,25-二羟基维生素D3 A环合成子26的另法合成
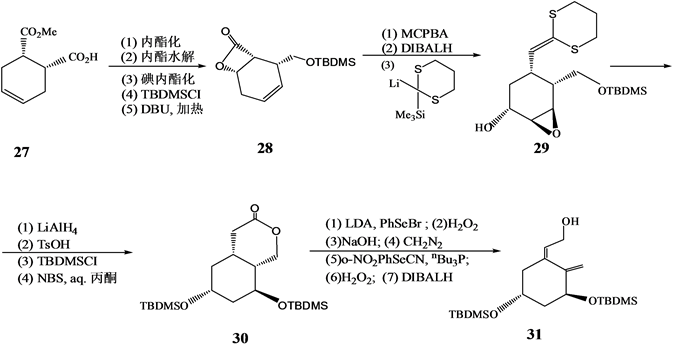
Scheme 7. Synthesis of 1α,25-dihydroxyvitamin D3 A ring synthon 31
Scheme 7. 1α,25-二羟基维生素D3 A环合成子31的另法合成

Scheme 8. Synthesis of 1α,25-dihydroxyvitamin D3 A ring synthon 37
Scheme 8. 1α,25-二羟基维生素D3 A环合成子37的另法合成
Okamula提出。它是通过A环炔烃与CD环烯三氟甲基磺酸酯的钯催化偶联反应制备。其中A环炔烃和CD环烯三氟甲基磺酸酯分别由(S)-香芹酮(S-Carvone)和Grundmann’s酮为起始原料,Grundmann’s酮通常由VD3或VD2臭氧化降解产物二醇38制得。
其合成路线如[18] (Scheme 9)。
莽草酸和奎尼酸是一种廉价的手性原料,它们也被用于维生素D类化合物A环的合成。Scheme 10给出的是Desmaele[19] 用莽草酸合成A环中间体47的路线。
从香芹酮出发,Mourin[20] 通过11步反应以10%的总收率合成了50(Scheme 11)。
Stepanenko和Wicha[21] 报道了(24S),25-二羟基维生素D的CD环合成子的对映体选择性合成。二酮51很容易通过酵母还原得到光学活性羟基酮(S)-52[22] ,因此羟基酮(S)-52用MCPBA氧化得到内酯(S)-53。内酯53开环,并制备亚丙基化合物54。54和LDA反应,得到相应的烯醇化物,然后用三甲基硅氯来终止反应。产物55进行两次Mukaiyama-Michael共轭加成,反应是55和56在TrSbCl6存在下,其后再和57反应,得到大部分组分是58的混合物,重结晶,用DIBALH处理,得到差向异构体二醇混合物,然后伯羟基用对甲苯磺酰氯酯化,硫醚氧化为砜,这个砜接着用过量LiALH4处理得到反式茚烷,然后脱掉保护基得到所需要的二醇(24S)-60(Scheme 12)。
3.2. 通过C6~C7键连接A环和CD环的合成策略
通过C6~C7键连接A环和CD环的合成策略如图2。
Vandewalle[23] 小组以环丙烷醛衍生物作为A环合成子,以烯溴衍生物作为CD环合成子,完成了临床试验治疗牛皮癣药物的TX522 70的合成(Scheme 13)。
Vandewalle小组[24] 报道了19-失碳-1α,25-二羟基VD3 3的合成,它是由25-羟基Grundemann’s酮衍生物66和一个顺式1,3,5-环己三醇71通过化学酶方法合成的A环前体合成子来完成的,其合成路线如
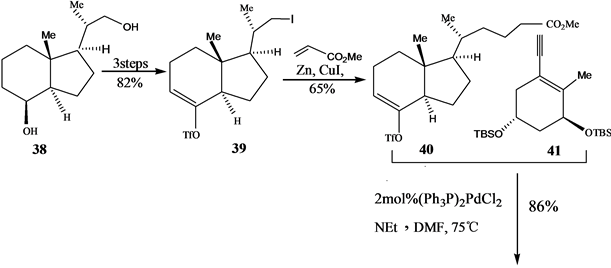

Scheme 9. Palladium catalyzed coupling united into 1α,25-dihydroxyvitamin D3 by A ring alkyne and CD cycloolefin three fluorinated methyl sulfonic acid ester
Scheme 9. A环炔烃与CD环烯三氟甲基磺酸酯的钯催化偶联合成1α,25-二羟基VD3

Scheme 10. Synthetic rout from shikimic acid to A ring synthon 47
Scheme 10. 由莽草酸合成A环中间体47的路线

Scheme 11. Synthetic rout from carvone to A ring synthon 50
Scheme 11. 由香芹酮合成A环中间体50的路线
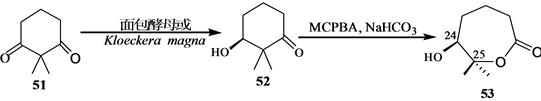
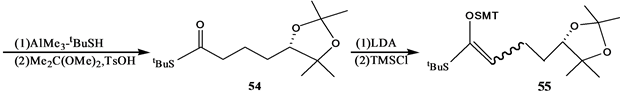

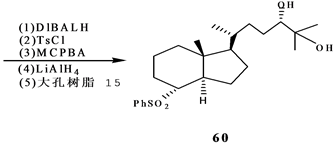
Scheme 12. Synthesis of 1α,25-dihydroxyvitamin D3 CD ring synthon
Scheme 12. 1α,25-二羟基VD3的CD环合成子的合成
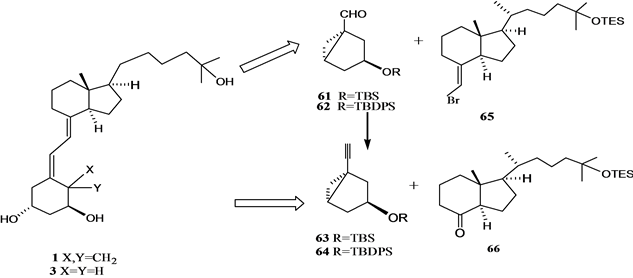
Figure 2. The retrosynthesis analysis of 1α,25-dihydroxy VD3 1 and 19-nor 1α, 25-dihydroxyVD3 3 by C6~C7 link
图2. 1α,25-二羟基VD3 1的和19-失碳-1α,25-二羟基VD3 3通过C6~C7键连接的逆合成分析
图(Scheme 14)。
吴勇[25] 以廉价易得的D-(-)奎尼酸为手性源,通过9步反应,有效合成19-失碳-1α,25-二羟基VD3 A环合成子炔64。作者[26] 以顺,顺,顺-(3,5-二羟基-4-乙基)环己基甲酸酯为原料,通过脂酶催化的不对称水解等8步完成了2-乙基取代的A环合成子83和84的合成,并与CD环的烯溴85反应完成了两种新的2-乙基19-失碳-1α,25-二羟基VD3类似物86和87的合成(Scheme 15)。陆群[27] 采用化学与生物转化相结合的方法合成了帕立骨化醇。
3.3. 烯炔的钯催化环化反应法——Trost法
Trost[28] 发展了一个利用烯炔的钯催化环化反应合成VD3的方法,其特点是通过CD环与烯炔反应,同时连接C6~C7键和C10~C15键从而实现1α,25-二羟基VD3的合成。其合成路线如下(Scheme 16)。最近,

Scheme 13. Synthesis of 1α,25-dihydroxyvitamin D3 analogue 70 by Vandewalle
Scheme 13. Vandewalle合成1α,25-二羟基维生素D3法类似物70的方法
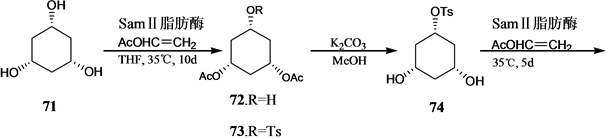
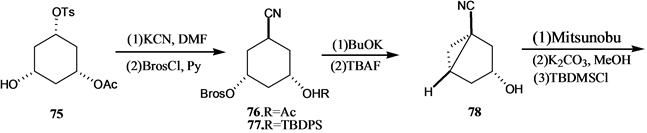
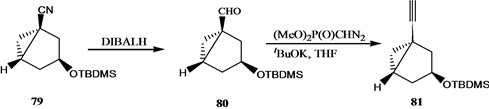
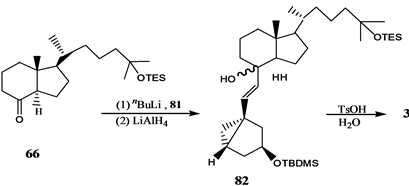
Scheme 14. Synthesis of 19-nor-1α,25-dihydroxyvitamin D3 3
Scheme 14. 19-失碳-1α,25-二羟基VD3 3的合成
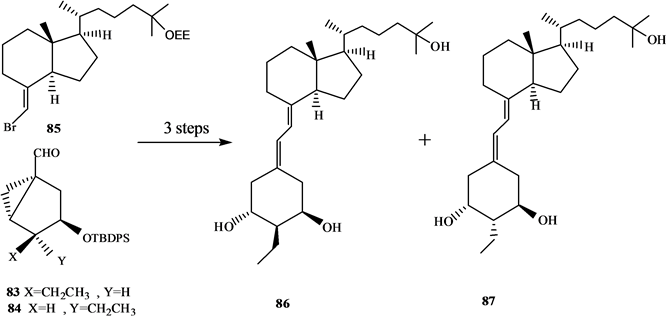
Scheme 15. Synthesis of 2-ethyl -19-nor-1α,25-dihydroxyvitamin D3 analogues
Scheme 15. 2-乙基-19-失碳-1α,25-二羟基VD3类似物的合成
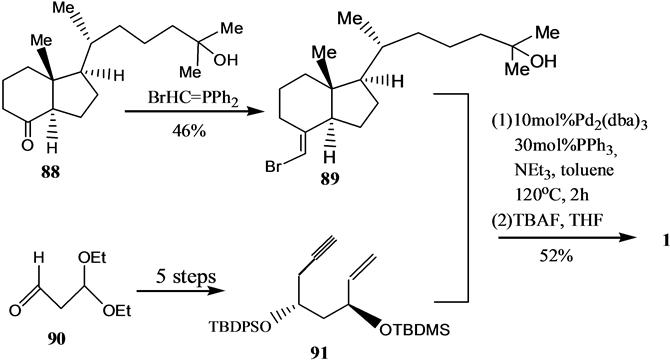
Scheme 16. Synthesis of VD3 by Palladium catalyzed olefin cyclization reaction
Scheme 16. 利用烯炔的钯催化环化反应合成VD3
Yoshitomo等[29] 用Torst策略合成了各种2α-取代的1α,25-二羟基VD3类似物。
4. 结语
自从1922年麦克勒姆(McCllum)发现维生素D,1927年温道斯(Windaus)推导出麦角甾醇是食物中维生素D的前体以后,早期人们仅仅将其视为一种维生素类物质,后来生物学表明:维生素D也是一种内源性激素物质。以往人们仅仅把维生素D作为钙和磷的调节剂,用其治疗小儿佝偻病、软骨病和骨质疏松症。1971年德卢卡(Deluca)、诺曼(Norman)和科迪塞克(Kodicek)三个小组几乎同时确认维生素D最终活性形式为1α,25-二羟基VD3。随后的研究表明,活性维生素D类化合物具有细胞诱导分化作用和阻止肿瘤细胞增生作用,可用于治疗肿瘤、牛皮癣和自身免疫疾病等。活性维生素D类化合物的化学和生物学研究引起人们高度重视。有机化学家通过各种方法合成了1α,25-二羟基VD3及其大量类似物。对活性VD3的合成至今仍以传统的液相反应为主。由于活性VD3的合成往往要经历很长的反应路线、多步操作及纯化等不足。目前,合成化学家们利用固相合成的高转化率、简单的洗涤式纯化过程等特点,开发出了用固相方式合成活性VD3的方法。如维生素D3衍生物Calcitriol的合成,可对A、C及D环进行完全方式的固相组装,而且A环增加了结构多样性修饰。例如,Xu等[30] 采用固相有机硒树脂并通过[2,3]σ迁移合成了Vitamin D类似物Deltanoid。近年来由于不对称合成的发展,使用新反应和新试剂,提高立体选择性和专一性,人们的注意力更多的转移到会聚合成的方法上。为了发展新型的老年疾病药物和抗肿瘤药物,人们除了继续寻找更经济、更实用的合成已知活性维生素D3类化合物的方法外,还需设计合成出更多更新的活性维生素D3类化合物。

NOTES
*通讯作者。