1. 引言
盐酸西那卡塞(Cinacalcet hydrochloride),化学名为N-[(1R)-1-(1-萘基)乙基]-3-[3-(三氟甲基)苯基]丙基胺盐酸盐,是由美国NPS Pharmaceuticals公司开发研究的第二代拟钙剂,于2004年首次在美国上市。临床上用于治疗进行透析的慢性肾病(CKD)患者的继发
性甲状旁腺功能亢进症及甲状旁腺肿瘤患者的高钙血症。本品的主要药理作用是降低Ca2+调定点,提高钙敏感受体对细胞外钙的敏感性,降低甲状旁腺素水平,使血清Ca2+浓度降低,从而产生一系列临床治疗作用。具有安全性高、耐受性好、服用方便等特点[1]。
盐酸西那卡塞的合成方法已有多篇文献报道,主要有以下七条合成路线:1) 以3-三氟甲基苯丙胺和1-萘乙酮为起始原料,在Ti(OiPr)4催化下进行缩合,经腈基硼氢化钠还原,最后用手性柱分离得到目标化合物[2]。该工艺第一步反应用到的Ti(OiPr)4价格较高,后处理非常困难,无水要求高;第二步中不饱和双键的还原采用氰基硼氢化钠,价格较贵,后处理较为麻烦;另外,目标产物需用手性色谱柱进行分离,只能少量制备,不适合工业化生产。2) 以3-三氟甲基苯丙醛和(R)-1-萘乙胺为起始原料,在Ti(OiPr)4催化下进行缩合反应,再经氰基硼氢化钠还原、成盐制得盐酸西那卡塞[3]。该工艺路线较为简便,但该路线的关键原料3-三氟甲基苯丙醛难以制备和购买,而且价格昂贵;同时该路线还使用了价格较贵的(R)-1-萘乙胺和Ti(OiPr)4,不利于降低成本。3) 以1-溴-3-三氟甲基苯为原料,通过Heck反应得到3-三氟甲基肉桂酸乙酯,经钯碳和氢化铝锂还原得到间三氟甲基苯丙醇,制成卤代物后,与(R)-1-萘乙胺发生亲核取代反应,最后成盐得到盐酸西那卡塞[4]。首先该工艺中3-三氟甲基肉桂酸乙酯的合成用到了丙烯酸乙酯,该试剂本身是己知的致癌物质,出于安全性考虑,在工业化生产过程中应该尽量避免使用;其次,Heck偶联需要使用较昂贵的钯催化剂,不利于降低成本。4) 以手性叔丁基硫酰胺、1-萘乙酮和3-三氟甲基苯甲醛为起始原料,合成两个侧链:叔丁基硫酰基保护的(R)-1-萘乙胺和间三氟甲基苯丙基卤代物,再经缩合、脱保护、成盐制得盐酸西那卡塞[5]。该工艺合成步骤长,手性原料价格高,需要使用催化剂Ti(OEt)4,强碱六甲基二硅基胺基锂(LiHMDS),成本较高;第一步反应时间太长(30 h),且需要在零下48℃反应,对设备要求较高,中间体分离难度较大,综合考虑,不适合大规模工业化生产。5) 以3-溴三氟甲苯与N-烯丙基-(R)-1-萘乙胺为起始原料在Pd或Ni催化下得到不饱和的西那卡塞中间体,再经钯碳还原、成盐后得到西那卡塞[6]。该工艺步骤较短,便于质量控制,但中间体N-烯丙基-(R)-1-萘乙胺制备难度大,不易购买,氮原子多数情况下需要用保护基进行保护,又增加了反应步骤,Heck偶联反应需要使用价格昂贵的金属催化剂,不利于工业化。6) 以(R)-1-萘乙胺和苯甲醛为起始原料,反应制得亚胺中间体,再与3-三氟甲基苯丙基溴在NMP存在下缩合生成亚胺盐,最后脱去苯甲醛、成盐得到盐酸西那卡塞[7]。该路线中,原料(R)-1-萘乙胺价
格较贵,亚胺中间体不稳定,不易储存,后处理复杂,用到二类溶剂甲苯,毒性大,不利于安全保护。7) 以3-三氟甲基苯丙酸为起始原料,制备成酰氯,与(R)-1-萘乙胺缩合,再经氢化铝锂或者硼烷等还原剂还原后成盐制得盐酸西那卡塞[8-10]。该工艺,步骤较短,条件温和,具有开发价值,但也存在原料(R)-1-萘乙胺不易得,生产成本较高,氢化铝锂作为还原剂不适于工业使用等缺点,有待进一步改进优化。
本文针对以上工艺路线的不足,通过大量文献调研和实验尝试,在方法7)的基础上进行了改进和优化,最终确定了一条原料易得、成本低廉、操作简便、各步分离纯化方便、收率较高、产品质量好、能够大规模工业化制备的盐酸西那卡塞的合成工艺。以1-萘乙酮为起始原料,还原胺化制得1-萘乙胺,经D-酒石酸拆分制得(R)-1-萘乙胺(5);将酰氯6与5缩合,制得(R)-N-(1-萘乙基)-3-(3-三氟甲基)苯丙酰胺(7),再经NaBH4-BF3·OEt2体系将酰胺还原,在5N HCl溶液中回流反应2 h,制得西那卡塞盐酸盐(1)。工艺路线见图1。
2. 实验部分
2.1. 主要仪器与试剂
Agilent1946A-MSD型质谱仪(ESI-MS);Bruker AV300型(300 Hz)核磁共振仪;岛津LC-15C高效液相色谱仪;WRS-1B数字熔点仪。1-萘乙酮(含量 ≥ 98%,南京多博化工有限公司),D-(-)-酒石酸(含量99.5%,阿拉丁试剂公司),3-三氟甲基苯丙酸(含量≥97%,山西津津化工有限公司)。所用试剂均为分析纯。
2.2. 合成方法
2.2.1. N-(1-萘乙基)甲酰胺的(3)的合成
向5 L三颈瓶中加入1-萘乙酮(500 g, 2.94 mol),甲酸铵(1480 g, 23.50 mol),缓慢加热至回流,回流分水反应5 h,冷却至室温,加入2 L二氯甲烷溶解后,倒入2 L水中,分层,水相用二氯甲烷萃取,合并有机层,分别用水,饱和氯化钠溶液洗涤,无水硫酸钠干燥,浓缩滤液,得类白色固体,粗品用石油醚/乙酸乙酯打浆精制,抽滤,洗涤滤饼,得白色粉末状固体,干燥得产品(3) 470 g,收率80.23%。mp:138.1℃~138.7℃。
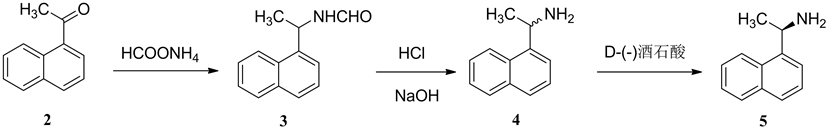
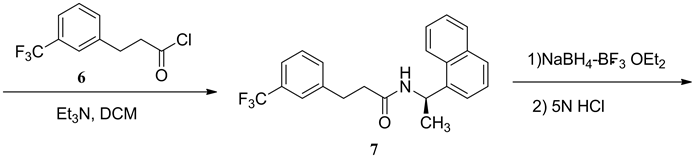
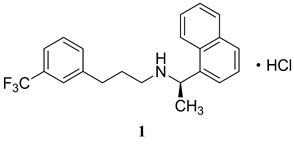
Figure 1. Synthesis of cinacalcet hydrochloride
图1. 盐酸西那卡塞的合成
1H NMR (300 MHz, CDCl3) δ 8.18 (1H, s,ArH), 8.10(1H, d, J = 8.1 Hz, ArH), 7.87(1H, d, J = 7.8 Hz, ArH), 7.81(1H, d, J = 7.9 Hz, ArH), 7.45-7.60(3H, m, ArH), 6.02(1H, q, J = 6.9 Hz, CH), 5.79(1H, s, CHO), 2.05(1H, d, J = 5.9 Hz, NH), 1.72(3H, d, J = 6.7 Hz, CH3)
2.2.2. 1-萘乙胺的(4)的合成
向5 L反应瓶中加入3(470 g, 2.36 mol),浓盐酸2 L,搅拌溶解,加热回流,溶液由浑浊逐渐变澄清,回流反应2 h后,冷却至室温,析出大量白色固体,抽滤,用乙酸乙酯洗涤得亮白色固体,直接加入5% NaOH溶液调节pH = 11,用乙酸乙酯萃取,合并有机层,饱和氯化钠溶液洗涤,无水硫酸钠干燥,抽滤,浓缩滤液得浅黄色透明油状物(4)343.50g,收率85%。
1H NMR (300 MHz, CDCl3) δ 8.17 (1H, d, J = 8.3 Hz, ArH), 7.95 - 7.85 (1H, m, ArH), 7.78 (1H, d, J = 8.2 Hz, ArH), 7.69 (1H, d, J = 7.1 Hz, ArH), 7.60 - 7.45 (3H, m, ArH), 4.99 (1H, q, J = 6.5 Hz, CH), 1.77 (2H, s, NH2), 1.59 (3H, d, J = 6.6 Hz, CH3).
2.2.3. (R)-1-萘乙胺(5)的合成
向5 L反应瓶中加入4(100 g, 583.98 mmol),加入250 ml甲醇搅拌溶解,缓慢加入D-酒石酸(87.65 g, 583.98 mmol),加毕析出大量白色固体,通过恒压滴
液漏斗滴加甲醇/水的混合溶剂2.5 L。加毕,加热搅拌溶解,全部溶解后停止加热,冷却至室温析晶,抽滤,乙酸乙酯洗涤滤饼得白色粉末状固体,干燥得71.50 g,光学纯度98.11%。用甲醇/水的混合溶剂重结晶得57.20 g,光学纯度99.9%,加入氢氧化钠溶液游离出(5) 30.30 g,拆分收率30.32%,e.e.值99.8%。
HPLC检测条件:色谱柱:C18,流动相:0.1%冰乙酸(三乙胺调节pH至5.0)-乙腈(79:21),流速:1.0 ml/min,柱温:40℃,检测波长:220 nm。
1H NMR (300 MHz, DMSO) δ 8.18 (1H, d, J = 8.0 Hz, ArH), 7.96 - 7.85 (1H, m, ArH), 7.74-7.78 (2H, m, ArH), 7.60 - 7.40 (3H, m, ArH), 4.83 (1H, q, J = 6.5 Hz, CH), 1.99 (2H, s,NH2), 1.39 (3H, d, J = 6.5 Hz,CH3).
2.2.4. (R)-N-(1-萘乙基)-3-(3-三氟甲基)苯丙酰胺(7) 的合成
将(R)-1-萘乙胺(164g, 957.72 mmol)加入2.5 L反应瓶中,用500 ml无水二氯甲烷搅拌至溶解,加入三乙胺(154.60 ml, 1.10 mol),室温搅拌10 min。取3-三氟甲基苯丙酰氯6(215.82 g, 912.11 mmol),溶于1L无水二氯甲烷,在冰浴下滴加入R-1-萘乙胺溶液中,30 min加毕,常温搅拌反应4~5 h。将反应液倒入2 L水中,用二氯甲烷萃取分液,合并二氯甲烷层,分别用2L 5%的氢氧化钠溶液、2 L × 2的饱和氯化钠溶液洗涤,无水硫酸钠干燥,抽滤,浓缩滤液得类白色固体,7的粗品371.40 g,于粗品中加入3 L混合溶剂(石油醚/乙酸乙酯)加热回流打浆2~3 h。冷却至室温,抽滤,洗涤滤饼得白色固体,干燥得7的精制品305.60 g,收率90.21%,HPLC纯度99.6%。mp:125.7℃~125.9℃
1H NMR (300 MHz, DMSO) δ 8.42 (1H, d, J = 7.9 Hz, ArH), 8.11-8.00 (1H, m, ArH), 7.98-7.87 (1H, m, ArH), 7.80 (1H, d, J = 6.7 Hz, ArH), 7.58-7.40 (7H, m, ArH), 5.73-5.60 (1H, m, CH), 2.93 (2H, t, J = 7.3 Hz, CH2), 2.57-2.40 (2H, t, J = 7.3 Hz, CH2), 1.43 (3H, d, J = 6.8 Hz, CH3).
2.2.5. 盐酸西那卡塞(1)的合成
向5 L茄形瓶中加入7(300 g, 807.75 mmol),加入2 L无水四氢呋喃搅拌溶解,加入NaBH4(61.11 g, 1.62 mol),冰浴降温,通过恒压滴液漏斗滴加NaBH4- BF3·OEt2溶液(204.40 ml, 1.62 mol),30 min加毕,加热回流反应4~5 h。反应完毕,冷却至室温,蒸除THF,加入2L 5%稀盐酸溶液淬灭,用乙酸乙酯萃取合并有机层,再用饱和氯化钠溶液洗涤,无水硫酸钠干燥,抽滤,浓缩滤液得浅黄色油状物西那卡塞341.40 g。加入1.5L5 N HCl,加热回流,搅拌反应2 h(捣碎结块固体),冷却至室温。砂芯漏斗抽滤,滤饼用水洗涤得白色固体,干燥,即得(1)293.20 g,收率92.36%,HPLC纯度99.34%,[α]D20 = −24.86˚ (c = 0.1 g/ml,无水乙醇),e.e. = 99%,文献值[α]D20 = −25˚ (c = 0.1 g/ml,无水乙醇)[11],mp:181.5℃~182℃(文献181℃~182℃)。
1H NMR (300 MHz, DMSO) δ 10.03 (1H, s, HCl), 9.35 (1H, s, NH), 8.25 (1H, d, J = 7.9 Hz, ArH), 8.01 (3H, dd, J = 15.4, 6.7 Hz, ArH), 7.68-7.42 (7H, m, ArH), 5.31 (1H, q, J = 5.0 Hz, CH), 2.96 (1H, m, CH2), 2.72 (3H, t, J = 7.5 Hz, CH2), 2.02 (2H, dd, J = 15.4, 7.7 Hz, CH2), 1.69 (3H, d, J = 6.6 Hz, CH3). MS(ESI(+)70 V) m/z 358.2[M + H]+。
取上述1的粗品280 g(HPLC纯度99.34%),加入500 ml异丙醇,加热搅拌溶解,冷却至室温,析出白色固体,用砂芯漏斗抽滤,滤饼用少量异丙醇洗涤,烘干得盐酸西那卡塞精制品233.50 g,收率83.4%,HPLC纯度99.9%,单个杂质均 < 0.1%。
HPLC检测条件:
色谱柱:Zorbax SB-Phenyl(4.6 × 250 mm,5 µm或类似极性)。
流动相:MPA:0.1%冰醋酸(用三乙胺调pH至5.5)-乙腈-甲醇(45:35; 20)。
MPB:乙腈-0.1%冰醋酸(用三乙胺调pH至5.5)(80:20)。
按下表进行梯度洗脱:

检测波长:220 nm,流速:1.0 ml/min,柱温:35℃,溶剂:水–乙腈(50:50)。
3. 结果与讨论
(R)-1-萘乙胺是合成盐酸西那卡塞的关键中间体,价格较高。文献报道的不对称合成法[12]成本较高,且很难得到光学纯度较高的产品。而使用酶[13]对消旋体进行拆分时,操作繁琐,不易进行。另外,目前报道的化学拆分剂如手性α-羟基萘乙酸、手性甘油醇衍生物、D-(+)-萘普生等价格较高。本工艺选用D-酒石酸作为拆分剂,甲醇/水的混合溶剂为拆分溶剂和重结晶溶剂,对1-萘乙胺进行拆分,收率30.3%,e.e.值99.8%。与文献报道的拆分方法相比,本工艺所选择的拆分剂廉价易得,拆分溶剂环保且可回收利用,降低了生产成本。
其中7的制备过程中,我们发现采用石油醚/乙酸乙酯混合溶剂打浆,可以除去大部分杂质,经过打浆精制后7的HPLC纯度达到99.6%左右,收率90%。西那卡塞的成盐,采用在稀盐酸中回流[14],析出白色固体状的产品,直接抽滤,水洗即可,相比在乙酸乙酯-盐酸溶液或者甲醇-盐酸溶液中成盐,简化了后处理操作。对于1的精制,我们发现采用异丙醇作为重结晶溶剂,精制效果明显高于甲醇/水、乙醇/水的体系,精制后的HPLC纯度达到99.9%。
4. 结论
本文所述的盐酸西那卡塞的合成路线是在现有文献报道基础上经过整合而成,我们对文献条件进行了改进和优化。以1-萘乙酮为起始原料,经6步反应制得盐酸西那卡塞,总收率14.38%(此合成路线总收率未经文献报道),目标产物经HPLC检测纯度达99.9%,单个杂质都在0.1%以下。本工艺具有操作简便、工艺稳定、条件温和、成本低廉、适合工业化生产等优点。

NOTES
*通讯作者。